Statistical mechanics
| Statistical Mechanics | ||||||||||
 |
||||||||||
Thermodynamics · Kinetic theory
|
||||||||||
| Classical mechanics | ||||||||||
History of classical mechanics · Timeline of classical mechanics
|
||||||||||
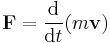
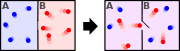
In physics, statistical mechanics (or statistical thermodynamics[1]) is the application of probability theory (which contains mathematical tools for dealing with large populations) to the study of the thermodynamic behavior of systems composed of a large number of particles. It provides a framework for relating the microscopic properties of individual atoms and molecules to the macroscopic or bulk properties of materials that can be observed in everyday life, therefore explaining thermodynamics as a natural result of statistics and mechanics (classical and quantum) at the microscopic level.
Statistical mechanics provides a molecular-level interpretation of macroscopic thermodynamic quantities such as work, heat, free energy, and entropy, allowing the thermodynamic properties of bulk materials to be related to the spectroscopic data of individual molecules. This ability to make macroscopic predictions based on microscopic properties is the main advantage of statistical mechanics over classical thermodynamics. Both theories are governed by the second law of thermodynamics through the medium of entropy. However, entropy in thermodynamics can only be known empirically, whereas in statistical mechanics, it is a function of the distribution of the system on its micro-states.
Statistical mechanics (or statistical thermodynamics) was initiated in 1870 with the work of Austrian physicist Ludwig Boltzmann, much of which was collectively published in Boltzmann's 1896 Lectures on Gas Theory.[2] Boltzmann's original papers on the statistical interpretation of thermodynamics, the H-theorem, transport theory, thermal equilibrium, the equation of state of gases, and similar subjects, occupy about 2,000 pages in the proceedings of the Vienna Academy and other societies. The term "statistical thermodynamics" was proposed for use by the American thermodynamicist and physical chemist J. Willard Gibbs in 1902. According to Gibbs, the term "statistical", in the context of mechanics, i.e. statistical mechanics, was first used by the Scottish physicist James Clerk Maxwell in 1871.
Contents |
Overview
The essential problem in statistical thermodynamics is to determine the distribution of a given amount of energy E over N identical systems.[3] The goal of statistical thermodynamics is to understand and to interpret the measurable macroscopic properties of materials in terms of the properties of their constituent particles and the interactions between them. This is done by connecting thermodynamic functions to quantum-mechanic equations. Two central quantities in statistical thermodynamics are the Boltzmann factor and the partition function.
Fundamentals
Central topics covered in statistical thermodynamics include:
- Microstates and configurations
- Boltzmann distribution law
- Partition function, Configuration integral or configurational partition function
- Thermodynamic equilibrium - thermal, mechanical, and chemical.
- Internal degrees of freedom - rotation, vibration, electronic excitation, etc.
- Heat capacity – Einstein solids, polyatomic gases, etc.
- Nernst heat theorem
- Fluctuations
- Gibbs paradox
- Degeneracy
Lastly, and most importantly, the formal definition of entropy of a thermodynamic system from a statistical perspective is called statistical entropy, and is defined as:
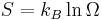
where
- kB is Boltzmann's constant 1.38066×10−23 J K−1 and
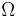 is the number of microstates corresponding to the observed thermodynamic macrostate.
is the number of microstates corresponding to the observed thermodynamic macrostate.
This equation is valid only if each microstate is equally accessible (each microstate has an equal probability of occurring).
Boltzmann Distribution
If the system is large the Boltzmann distribution could be used (the Boltzmann distribution is an approximate result)
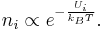
This can now be used with 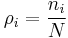 :
:
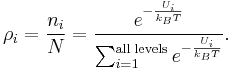
History
In 1738, Swiss physicist and mathematician Daniel Bernoulli published Hydrodynamica which laid the basis for the kinetic theory of gases. In this work, Bernoulli posited the argument, still used to this day, that gases consist of great numbers of molecules moving in all directions, that their impact on a surface causes the gas pressure that we feel, and that what we experience as heat is simply the kinetic energy of their motion.
In 1859, after reading a paper on the diffusion of molecules by Rudolf Clausius, Scottish physicist James Clerk Maxwell formulated the Maxwell distribution of molecular velocities, which gave the proportion of molecules having a certain velocity in a specific range. This was the first-ever statistical law in physics.[4] Five years later, in 1864, Ludwig Boltzmann, a young student in Vienna, came across Maxwell’s paper and was so inspired by it that he spent much of his long and distinguished life developing the subject further.
Hence, the foundations of statistical thermodynamics were laid down in the late 1800s by those such as Maxwell, Boltzmann, Max Planck, Clausius, and Willard Gibbs who began to apply statistical and quantum atomic theory to ideal gas bodies. Predominantly, however, it was Maxwell and Boltzmann, working independently, who reached similar conclusions as to the statistical nature of gaseous bodies. Yet, one must consider Boltzmann to be the "father" of statistical thermodynamics with his 1875 derivation of the relationship between entropy S and multiplicity Ω, the number of microscopic arrangements (microstates) producing the same macroscopic state (macrostate) for a particular system.[5]
Fundamental postulate
The fundamental postulate in statistical mechanics (also known as the equal a priori probability postulate) is the following:
- Given an isolated system in equilibrium, it is found with equal probability in each of its accessible microstates.
This postulate is a fundamental assumption in statistical mechanics - it states that a system in equilibrium does not have any preference for any of its available microstates. Given Ω microstates at a particular energy, the probability of finding the system in a particular microstate is p = 1/Ω.
This postulate is necessary because it allows one to conclude that for a system at equilibrium, the thermodynamic state (macrostate) which could result from the largest number of microstates is also the most probable macrostate of the system.
The postulate is justified in part, for classical systems, by Liouville's theorem (Hamiltonian), which shows that if the distribution of system points through accessible phase space is uniform at some time, it remains so at later times.
Similar justification for a discrete system is provided by the mechanism of detailed balance.
This allows for the definition of the information function (in the context of information theory):
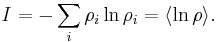
When all the probabilities (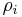 ) are equal, I is maximal, and we have minimal information about the system. When our information is maximal (i.e., one rho is equal to one and the rest to zero, such that we know what state the system is in), the function is minimal.
) are equal, I is maximal, and we have minimal information about the system. When our information is maximal (i.e., one rho is equal to one and the rest to zero, such that we know what state the system is in), the function is minimal.
This "information function" is the same as the reduced entropic function in thermodynamics.
Statistical ensembles
- See also: Statistical ensemble
Microcanonical ensemble
In microcanonical ensemble N, V and E are fixed. Since the second law of thermodynamics applies to isolated systems, the first case investigated will correspond to this case. The Microcanonical ensemble describes an isolated system.
The entropy of such a system can only increase, so that the maximum of its entropy corresponds to an equilibrium state for the system.
Because an isolated system keeps a constant energy, the total energy of the system does not fluctuate. Thus, the system can access only those of its micro-states that correspond to a given value E of the energy. The internal energy of the system is then strictly equal to its energy.
Let us call 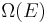 the number of micro-states corresponding to this value of the system's energy. The macroscopic state of maximal entropy for the system is the one in which all micro-states are equally likely to occur, with probability
the number of micro-states corresponding to this value of the system's energy. The macroscopic state of maximal entropy for the system is the one in which all micro-states are equally likely to occur, with probability 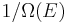 , during the system's fluctuations.
, during the system's fluctuations.
-
- where
 is the system entropy, and
is the system entropy, and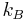 is Boltzmann's constant.
is Boltzmann's constant.
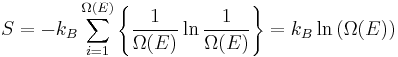
Canonical ensemble
In canonical ensemble N, V and T are fixed. Invoking the concept of the canonical ensemble, it is possible to derive the probability 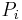 that a macroscopic system in thermal equilibrium with its environment, will be in a given microstate with energy
that a macroscopic system in thermal equilibrium with its environment, will be in a given microstate with energy 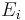 according to the Boltzmann distribution:
according to the Boltzmann distribution:
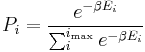
- where
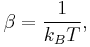
The temperature  arises from the fact that the system is in thermal equilibrium with its environment. The probabilities of the various microstates must add to one, and the normalization factor in the denominator is the canonical partition function:
arises from the fact that the system is in thermal equilibrium with its environment. The probabilities of the various microstates must add to one, and the normalization factor in the denominator is the canonical partition function:
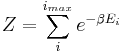
where is the energy of the  th microstate of the system. The partition function is a measure of the number of states accessible to the system at a given temperature. The article canonical ensemble contains a derivation of Boltzmann's factor and the form of the partition function from first principles.
th microstate of the system. The partition function is a measure of the number of states accessible to the system at a given temperature. The article canonical ensemble contains a derivation of Boltzmann's factor and the form of the partition function from first principles.
To sum up, the probability of finding a system at temperature in a particular state with energy is
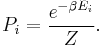
Thermodynamic Connection
The partition function can be used to find the expected (average) value of any microscopic property of the system, which can then be related to macroscopic variables. For instance, the expected value of the microscopic energy  is interpreted as the microscopic definition of the thermodynamic variable internal energy
is interpreted as the microscopic definition of the thermodynamic variable internal energy  , and can be obtained by taking the derivative of the partition function with respect to the temperature. Indeed,
, and can be obtained by taking the derivative of the partition function with respect to the temperature. Indeed,
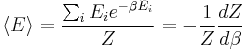
implies, together with the interpretation of 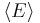 as , the following microscopic definition of internal energy:
as , the following microscopic definition of internal energy:
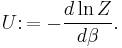
The entropy can be calculated by (see Shannon entropy)
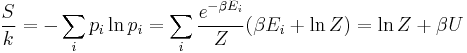
which implies that
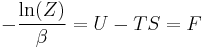
is the free energy of the system or in other words,
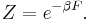
Having microscopic expressions for the basic thermodynamic potentials (internal energy), (entropy) and  (free energy) is sufficient to derive expressions for other thermodynamic quantities. The basic strategy is as follows. There may be an intensive or extensive quantity that enters explicitly in the expression for the microscopic energy , for instance magnetic field (intensive) or volume (extensive). Then, the conjugate thermodynamic variables are derivatives of the internal energy. The macroscopic magnetization (extensive) is the derivative of with respect to the (intensive) magnetic field, and the pressure (intensive) is the derivative of with respect to volume (extensive).
(free energy) is sufficient to derive expressions for other thermodynamic quantities. The basic strategy is as follows. There may be an intensive or extensive quantity that enters explicitly in the expression for the microscopic energy , for instance magnetic field (intensive) or volume (extensive). Then, the conjugate thermodynamic variables are derivatives of the internal energy. The macroscopic magnetization (extensive) is the derivative of with respect to the (intensive) magnetic field, and the pressure (intensive) is the derivative of with respect to volume (extensive).
The treatment in this section assumes no exchange of matter (i.e. fixed mass and fixed particle numbers). However, the volume of the system is variable which means the density is also variable.
This probability can be used to find the average value, which corresponds to the macroscopic value, of any property,  , that depends on the energetic state of the system by using the formula:
, that depends on the energetic state of the system by using the formula:
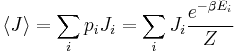
where 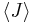 is the average value of property . This equation can be applied to the internal energy, :
is the average value of property . This equation can be applied to the internal energy, :
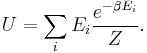
Subsequently, these equations can be combined with known thermodynamic relationships between and 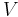 to arrive at an expression for pressure in terms of only temperature, volume and the partition function. Similar relationships in terms of the partition function can be derived for other thermodynamic properties as shown in the following table; see also the detailed explanation in configuration integral.
to arrive at an expression for pressure in terms of only temperature, volume and the partition function. Similar relationships in terms of the partition function can be derived for other thermodynamic properties as shown in the following table; see also the detailed explanation in configuration integral.
| Helmholtz free energy: | 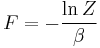 |
|---|---|
| Internal energy: | 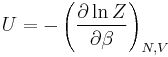 |
| Pressure: | 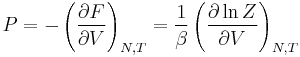 |
| Entropy: | 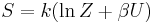 |
| Gibbs free energy: | 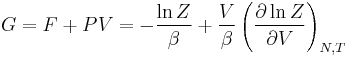 |
| Enthalpy: | 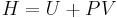 |
| Constant volume heat capacity: | 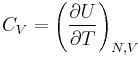 |
| Constant pressure heat capacity: | 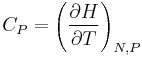 |
| Chemical potential: | 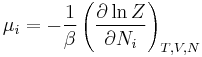 |
To clarify, this is not a grand canonical ensemble.
It is often useful to consider the energy of a given molecule to be distributed among a number of modes. For example, translational energy refers to that portion of energy associated with the motion of the center of mass of the molecule. Configurational energy refers to that portion of energy associated with the various attractive and repulsive forces between molecules in a system. The other modes are all considered to be internal to each molecule. They include rotational, vibrational, electronic and nuclear modes. If we assume that each mode is independent (a questionable assumption) the total energy can be expressed as the sum of each of the components:
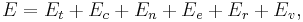
where the subscripts  ,
,  ,
, 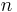 ,
, 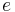 ,
,  , and
, and 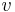 correspond to translational, configurational, nuclear, electronic, rotational and vibrational modes, respectively. The relationship in this equation can be substituted into the very first equation to give:
correspond to translational, configurational, nuclear, electronic, rotational and vibrational modes, respectively. The relationship in this equation can be substituted into the very first equation to give:
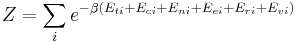
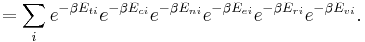
If we can assume all these modes are completely uncoupled and uncorrelated, so all these factors are in a probability sense completely independent, then
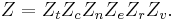
Thus a partition function can be defined for each mode. Simple expressions have been derived relating each of the various modes to various measurable molecular properties, such as the characteristic rotational or vibrational frequencies.
Expressions for the various molecular partition functions are shown in the following table.
| Nuclear | 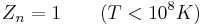 |
|---|---|
| Electronic | 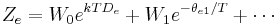 |
| Vibrational | 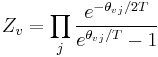 |
| Rotational (linear) | 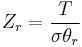 |
| Rotational (non-linear) | 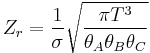 |
| Translational | 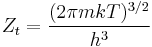 |
| Configurational (ideal gas) | 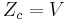 |
These equations can be combined with those in the first table to determine the contribution of a particular energy mode to a thermodynamic property. For example the "rotational pressure" could be determined in this manner. The total pressure could be found by summing the pressure contributions from all of the individual modes, i.e.:
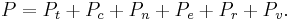
Grand canonical ensemble
In grand canonical ensemble , and chemical potential are fixed. If the system under study is an open system, (matter can be exchanged), but particle number is not conserved, we would have to introduce chemical potentials, μj, j = 1,...,n and replace the canonical partition function with the grand canonical partition function:
![\Xi(V,T,\mu) = \sum_i \exp\left(\beta \left[\sum_{j=1}^n \mu_j N_{ij}-E_i\right ]\right)](/2010-wikipedia_en_wp1-0.8_orig_2010-12/I/fa4a0a546cb56b26e20a56be1480d439.png)
where Nij is the number of jth species particles in the ith configuration. Sometimes, we also have other variables to add to the partition function, one corresponding to each conserved quantity. Most of them, however, can be safely interpreted as chemical potentials. In most condensed matter systems, things are nonrelativistic and mass is conserved. However, most condensed matter systems of interest also conserve particle number approximately (metastably) and the mass (nonrelativistically) is none other than the sum of the number of each type of particle times its mass. Mass is inversely related to density, which is the conjugate variable to pressure. For the rest of this article, we will ignore this complication and pretend chemical potentials don't matter. See grand canonical ensemble.
Let's rework everything using a grand canonical ensemble this time. The volume is left fixed and does not figure in at all in this treatment. As before, j is the index for those particles of species j and i is the index for microstate i:
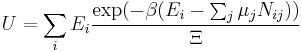
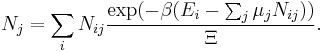
| Grand potential: | 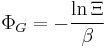 |
|---|---|
| Internal energy: | 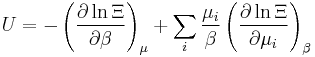 |
| Particle number: | 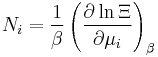 |
| Entropy: | 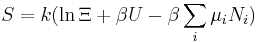 |
| Helmholtz free energy: | 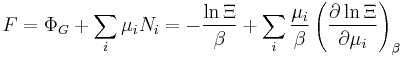 |
Equivalence between descriptions at the thermodynamic limit
All of the above descriptions differ in the way they allow the given system to fluctuate between its configurations.
In the micro-canonical ensemble, the system exchanges no energy with the outside world, and is therefore not subject to energy fluctuations; in the canonical ensemble, the system is free to exchange energy with the outside in the form of heat.
In the thermodynamic limit, which is the limit of large systems, fluctuations become negligible, so that all these descriptions converge to the same description. In other words, the macroscopic behavior of a system does not depend on the particular ensemble used for its description.
Given these considerations, the best ensemble to choose for the calculation of the properties of a macroscopic system is that ensemble which allows the result to be derived most easily.
Random walks
The study of long chain polymers has been a source of problems within the realms of statistical mechanics since about the 1950s. One of the reasons however that scientists were interested in their study is that the equations governing the behaviour of a polymer chain were independent of the chain chemistry. What is more, the governing equation turns out to be a random (diffusive) walk in space. Indeed, the Schrödinger equation is itself a diffusion equation in imaginary time, 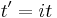 .
.
Random walks in time
The first example of a random walk is one in space, whereby a particle undergoes a random motion due to external forces in its surrounding medium. A typical example would be a pollen grain in a beaker of water. If one could somehow "dye" the path the pollen grain has taken, the path observed is defined as a random walk.
Consider a toy problem, of a train moving along a 1D track in the x-direction. Suppose that the train moves either a distance of + or - a fixed distance b, depending on whether a coin lands heads or tails when flipped. Lets start by considering the statistics of the steps the toy train takes (where 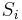 is the ith step taken):
is the ith step taken):
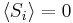 ; due to a priori equal probabilities
; due to a priori equal probabilities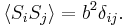
The second quantity is known as the correlation function. The delta is the kronecker delta which tells us that if the indices i and j are different, then the result is 0, but if i = j then the kronecker delta is 1, so the correlation function returns a value of 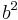 . This makes sense, because if i = j then we are considering the same step. Rather trivially then it can be shown that the average displacement of the train on the x-axis is 0;
. This makes sense, because if i = j then we are considering the same step. Rather trivially then it can be shown that the average displacement of the train on the x-axis is 0;
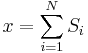
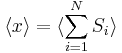
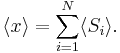
As stated 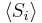 is 0, so the sum of 0 is still 0. It can also be shown, using the same method demonstrated above, to calculate the root mean square value of problem. The result of this calculation is given below
is 0, so the sum of 0 is still 0. It can also be shown, using the same method demonstrated above, to calculate the root mean square value of problem. The result of this calculation is given below
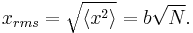
From the diffusion equation it can be shown that the distance a diffusing particle moves in a media is proportional to the root of the time the system has been diffusing for, where the proportionality constant is the root of the diffusion constant. The above relation, although cosmetically different reveals similar physics, where N is simply the number of steps moved (is loosely connected with time) and b is the characteristic step length. As a consequence we can consider diffusion as a random walk process.
Random walks in space
Random walks in space can be thought of as snapshots of the path taken by a random walker in time. One such example is the spatial configuration of long chain polymers.
There are two types of random walk in space: self-avoiding random walks, where the links of the polymer chain interact and do not overlap in space, and pure random walks, where the links of the polymer chain are non-interacting and links are free to lie on top of one another. The former type is most applicable to physical systems, but their solutions are harder to get at from first principles.
By considering a freely jointed, non-interacting polymer chain, the end-to-end vector is 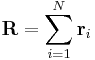 where
where 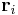 is the vector position of the i-th link in the chain. As a result of the central limit theorem, if N >> 1 then we expect a Gaussian distribution for the end-to-end vector. We can also make statements of the statistics of the links themselves;
is the vector position of the i-th link in the chain. As a result of the central limit theorem, if N >> 1 then we expect a Gaussian distribution for the end-to-end vector. We can also make statements of the statistics of the links themselves;
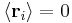 ; by the isotropy of space
; by the isotropy of space
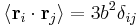 ; all the links in the chain are uncorrelated with one another
; all the links in the chain are uncorrelated with one another
Using the statistics of the individual links, it is easily shown that 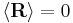 and
and 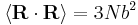 . Notice this last result is the same as that found for random walks in time.
. Notice this last result is the same as that found for random walks in time.
Assuming, as stated, that that distribution of end-to-end vectors for a very large number of identical polymer chains is gaussian, the probability distribution has the following form
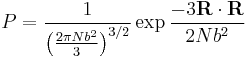
What use is this to us? Recall that according to the principle of equally likely a priori probabilities, the number of microstates, Ω, at some physical value is directly proportional to the probability distribution at that physical value, viz;
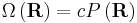
where c is an arbitrary proportionality constant. Given our distribution function, there is a maxima corresponding to 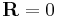 . Physically this amounts to there being more microstates which have an end-to-end vector of 0 than any other microstate. Now by considering
. Physically this amounts to there being more microstates which have an end-to-end vector of 0 than any other microstate. Now by considering
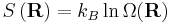
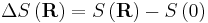
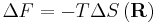
where F is the Helmholtz free energy it is trivial to show that
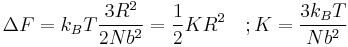
A Hookian spring!
This result is known as the Entropic Spring Result and amounts to saying that upon stretching a polymer chain you are doing work on the system to drag it away from its (preferred) equilibrium state. An example of this is a common elastic band, composed of long chain (rubber) polymers. By stretching the elastic band you are doing work on the system and the band behaves like a conventional spring. What is particularly astonishing about this result however, is that the work done in stretching the polymer chain can be related entirely to the change in entropy of the system as a result of the stretching.
Classical thermodynamics vs. statistical thermodynamics
As an example, from a classical thermodynamics point of view one might ask what is it about a thermodynamic system of gas molecules, such as ammonia NH3, that determines the free energy characteristic of that compound? Classical thermodynamics does not provide the answer. If, for example, we were given spectroscopic data, of this body of gas molecules, such as bond length, bond angle, bond rotation, and flexibility of the bonds in NH3 we should see that the free energy could not be other than it is. To prove this true, we need to bridge the gap between the microscopic realm of atoms and molecules and the macroscopic realm of classical thermodynamics. From physics, statistical mechanics provides such a bridge by teaching us how to conceive of a thermodynamic system as an assembly of units. More specifically, it demonstrates how the thermodynamic parameters of a system, such as temperature and pressure, are interpretable in terms of the parameters descriptive of such constituent atoms and molecules.[6]
In a bounded system, the crucial characteristic of these microscopic units is that their energies are quantized. That is, where the energies accessible to a macroscopic system form a virtual continuum of possibilities, the energies open to any of its submicroscopic components are limited to a discontinuous set of alternatives associated with integral values of some quantum number.
See also
- Chemical thermodynamics
- Configuration entropy
- Dangerously irrelevant
- Paul Ehrenfest
- Equilibrium thermodynamics
- Fluctuation dissipation theorem
- Important Publications in Statistical Mechanics
- Ising Model
- Mean field theory
- Nanomechanics
- Non-equilibrium thermodynamics
- Quantum thermodynamics
- Statistical physics
- Thermochemistry
- Widom insertion method
- Monte Carlo method
- Molecular modelling
| Maxwell Boltzmann | Bose-Einstein | Fermi-Dirac | |
|---|---|---|---|
| Particle | Boson | Fermion | |
| Statistics |
Partition function |
||
| Statistics |
Maxwell-Boltzmann statistics |
Bose-Einstein statistics | Fermi-Dirac statistics |
| Thomas-Fermi approximation |
gas in a box gas in a harmonic trap |
||
| Gas | Ideal gas |
Bose gas |
Fermi gas |
| Chemical Equilibrium |
Classical Chemical equilibrium | ||
Notes
- ↑ The terms "Statistical mechanics" and "statistical thermodynamics" are used interchangeably. "Statistical physics" is a broader term which includes statistical mechanics, but is sometimes also used as a synonym for statistical mechanics
- ↑ On history of fundamentals of statistical thermodynamics (section 1.2)
- ↑ Schrodinger, Erwin (1946). Statistical Thermodynamics. Dover Publications, Inc.. ISBN 0-486-66101-6. OCLC 20056858.
- ↑ Mahon, Basil (2003). The Man Who Changed Everything – the Life of James Clerk Maxwell. Hoboken, NJ: Wiley. ISBN 0-470-86171-1. OCLC 62045217 52358254 62045217.
- ↑ Perrot, Pierre (1998). A to Z of Thermodynamics. Oxford University Press. ISBN 0-19-856552-6. OCLC 38073404 123283342 38073404.
- ↑ Nash, Leonard K. (1974). Elements of Statistical Thermodynamics, 2nd Ed.. Dover Publications, Inc.. ISBN 0-486-44978-5. OCLC 61513215.
References
- Chandler, David (1987). Introduction to Modern Statistical Mechanics. Oxford University Press. ISBN 0-19-504277-8. OCLC 13946448.
- Huang, Kerson (1990). Statistical Mechanics. Wiley, John & Sons. ISBN 0-471-81518-7. OCLC 15017884.
- Kittel, Charles; Herbert Kroemer (1980). Thermal Physics (2nd ed.). San Francisco: W.H. Freeman. ISBN 0716710889. OCLC 5171399.
- McQuarrie, Donald (2000). Statistical Mechanics (2nd rev. Ed.). University Science Books. ISBN 1-891389-15-7. OCLC 43370175.
- Dill, Ken; Bromberg, Sarina (2003). Molecular Driving Forces. Garland Science. ISBN 0-8153-2051-5. OCLC 47915710.
- List of notable textbooks in statistical mechanics
External links
- Philosophy of Statistical Mechanics article by Lawrence Sklar for the Stanford Encyclopedia of Philosophy.
- Sklogwiki - Thermodynamics, statistical mechanics, and the computer simulation of materials. SklogWiki is particularly orientated towards liquids and soft condensed matter.
- Statistical Thermodynamics - Historical Timeline
- Thermodynamics and Statistical Mechanics by Richard Fitzpatrick
Further reading
- Ben-Naim, Arieh (2007). Statistical Thermodynamics Based on Information. ISBN 978-981-270-707-9
- Boltzmann, Ludwig; and Dieter Flamm (2000). Entropie und Wahrscheinlichkeit. ISBN 978-3817132867
- Boltzmann, Ludwig (1896, 1898). [Lectures on gas theory]. New York: Dover. ISBN 0486684555. OCLC 31434905. translated by Stephen G. Brush (1964) Berkeley: University of California Press; (1995) New York: Dover ISBN 0-486-68455-5
- Gibbs, J. Willard (1981) [1902]. Elementary principles in statistical mechanics. Woodbridge, Connecticut: Ox Bow Press. ISBN 0-918024-20-X.
- Landau, Lev Davidovich; and Lifshitz, Evgeny Mikhailovich (1980) [1976]. Statistical Physics. 5 (3 ed.). Oxford: Pergamon Press. ISBN 0-7506-3372-7. Translated by J.B. Sykes and M.J. Kearsley
- Reichl, Linda E (1998) [1980]. A modern course in statistical physics (2 ed.). Chichester: Wiley. ISBN 0-471-59520-9.
|
|||||
|
||||||||||||||||||||||||||